

Introduction
Dans ce chapitre, je vous propose d'accompagner la lumière, ou du moins un faisceau lumineux, au cours de son trajet. Cette lumière va se propager dans le vide puis elle va rencontrer des milieux et c'est à ce moment là que les choses ont devenir intéressantes...Que se passe-t-il quand la lumière traverse une vitre ou de l'eau est-ce la même chose? Qu'est ce qui change dans la lumière? Quand vous saurez répondre à ces questions, vous saurez ce qu'est la spectroscopie des rayonnements.I. Spectre et propagation
1. Rappel sur le spectre
La lumière, nous l'avons vu peut être décrite comme une onde ou comme des particules, quelque soit la vision que vous adopterez, le faisceau lumineux que nous observons correspond à la propagation de plusieurs longueurs d'ondes : il s'agit d'un faisceau polychromatique. En toute rigueur, le spectre d'une source est le graphe donnant le flux lumineux en fonction de la longueur d'onde.

2. Interaction matière rayonnement
Le faisceau lumineux arrive maintenant sur la matière, pour fixer les idées, du verre, en réalité peu importe la nature de la matière, tant que l'échantillon considéré est transparent. De la lumière arrive sur la matière et de la lumière en ressort, la première chose qui change c'est la quantité de lumière, le nombre de photons qui vont ressortir, c'est logique non? En réalité plusieurs processus interviennent mais tous aboutissent aux résultat suivant : lors de la traversée de la matière, le nombre de photon diminue et donc, l'énergie du faisceau lumineux diminue. Cette diminution d'énergie se doit d'être quantifiée, et pour cela, nous devons nous intéresser au flux lumineux . Pour faire simple, considérons une fine tranche de matière d'épaisseur très faible, nous la noterons dx. Cette tranche va provoquer une diminution du flux lumineux, et on peut supposer que cette diminution du flux est proportionnelle à l'épaisseur dx, de plus la diminution du flux est fonction du flux incident sur cette tranche (c'est un peu comme une éponge, si vous mettez 1L d'eau ou 1mL la capacité d'absorption de l'éponge n'est pas la même!!). On peut donc écrire la relation suivante entre flux lumineux arrivant sur la tranche et le flux lumineux qui en sort :
. Pour faire simple, considérons une fine tranche de matière d'épaisseur très faible, nous la noterons dx. Cette tranche va provoquer une diminution du flux lumineux, et on peut supposer que cette diminution du flux est proportionnelle à l'épaisseur dx, de plus la diminution du flux est fonction du flux incident sur cette tranche (c'est un peu comme une éponge, si vous mettez 1L d'eau ou 1mL la capacité d'absorption de l'éponge n'est pas la même!!). On peut donc écrire la relation suivante entre flux lumineux arrivant sur la tranche et le flux lumineux qui en sort :


 est appelé coefficient d'absorption linéique, il dépend de la nature du matériau traversé, en effet vous vous doutez bien que si l'absorption due à du verre n'est pas la même que celle due au plomb ! Mais ce coefficient dépend d'un autre paramètre très important pour nous: le coefficient d'absorption linéique dépend de l'énergie du faisceau incident et c'est cet dépendance qui va nous permettre de poser les bases de la spectroscopie...
est appelé coefficient d'absorption linéique, il dépend de la nature du matériau traversé, en effet vous vous doutez bien que si l'absorption due à du verre n'est pas la même que celle due au plomb ! Mais ce coefficient dépend d'un autre paramètre très important pour nous: le coefficient d'absorption linéique dépend de l'énergie du faisceau incident et c'est cet dépendance qui va nous permettre de poser les bases de la spectroscopie...
3. Les bases de la spectroscopie classique
La dernière remarque du paragraphe précédent nous mène sur la piste de l'énergie d'un faisceau lumineux. Cette énergie est fonction bien sur du nombre de photons ou du flux mais elle est aussi fonction de la longueur d'onde... En effet en adoptant un modèle corpusculaire, on décrit la lumière comme un ensemble de particules sans masse et d'énergie : ce sont les photons.
En face de ces photons, il y a la matière constituée d'un ensemble d'atomes, d'ions ou de molécules. Dans ces édifices, les électrons sont répartis sur des niveaux énergétiques, le plus souvent discret mais parfois rassemblés sous forme de 'bandes d'énergie'. Si on communique à ces électrons la quantité d'énergie adéquate, ils vont passer d'un niveau énergétique à un autre, mais cette transition n'est possible que si la quantité d'énergie qu'on fournit à l'électron correspond à un écart énergétique entre deux niveaux d'énergie, nous noterons
: ce sont les photons.
En face de ces photons, il y a la matière constituée d'un ensemble d'atomes, d'ions ou de molécules. Dans ces édifices, les électrons sont répartis sur des niveaux énergétiques, le plus souvent discret mais parfois rassemblés sous forme de 'bandes d'énergie'. Si on communique à ces électrons la quantité d'énergie adéquate, ils vont passer d'un niveau énergétique à un autre, mais cette transition n'est possible que si la quantité d'énergie qu'on fournit à l'électron correspond à un écart énergétique entre deux niveaux d'énergie, nous noterons  cet écart.
Lorsque les photons rencontrent la matière que va-t-il réellement se passer? Eh bien à chaque fois qu'un photon tombe sur un édifice qui veut bien de son énergie, c'est à dire à chaque fois que
cet écart.
Lorsque les photons rencontrent la matière que va-t-il réellement se passer? Eh bien à chaque fois qu'un photon tombe sur un édifice qui veut bien de son énergie, c'est à dire à chaque fois que  le photon va être absorbé par un électron qui va changer de niveau d'énergie. En revanche si au cours de sa traversée, le photon ne rencontre aucun édifice qui accepte son énergie, alors il traversera la matière sans être absorbé! Nous voici donc prêt à faire de la spectroscopie, en effet si on arrive à déceler les photons qui ont été absorbés et ceux qui sont passés sans encombres, on saura quels
le photon va être absorbé par un électron qui va changer de niveau d'énergie. En revanche si au cours de sa traversée, le photon ne rencontre aucun édifice qui accepte son énergie, alors il traversera la matière sans être absorbé! Nous voici donc prêt à faire de la spectroscopie, en effet si on arrive à déceler les photons qui ont été absorbés et ceux qui sont passés sans encombres, on saura quels  il y a dans la matière, or l'écart énergétique entre niveaux d'énergie est propre à chaque édifice... on peut donc savoir quels édifices ont été rencontrés en observant quels photons sont passés et quels photons sont arrêtés.
il y a dans la matière, or l'écart énergétique entre niveaux d'énergie est propre à chaque édifice... on peut donc savoir quels édifices ont été rencontrés en observant quels photons sont passés et quels photons sont arrêtés.L'idée de base est posée, pour la réaliser nous devrons être capable de faire une étude longueur d'onde par longueur d'onde pour cela, il nous faudra un système dispersif...
II. Le prisme
1. Description
Le premier système dispersif nous a été découvert par un certain Isaac Newton dans son laboratoire de Londres. Il s'agit d'une pièce de verre d'indice optique n, en forme de pyramide à base rectangle comme le montre la figure suivante.
Le prisme doit donc susciter votre plus grand intérêt, il permet d'obtenir des faisceaux monochromatiques, c'est parfait pour la spectroscopie, il suffit de les faire traverser tour à tour la matière et d'observer quels sont ceux qui sont absorbés! On en déduira les édifices présents dans la matière!! Reste donc à savoir comment placer le prisme et comment récupérer les faisceaux monochromatiques un par un...
1. Formules du prisme
La première chose que vous constaterez si vous êtes amenés à utiliser un prisme, c'est que le prisme dévie la lumière. Il convient donc de définir la déviation $D$ du prisme comme l'angle orienté du rayon incident vers le rayon émergent, la figure suivante illustre cette définition.
Première relation
L'application des lois de Descartes sur la réfraction au point I, permet décrire :
Deuxième relation
La seconde relation s'obtient aussi aisément que la première mais en se plaçant cette fois-ci au point J.
Troisième relation
Ecrivons que la somme des angles dans le triangle AIJ est égale à :
:






2. Déviation minimale
Le prisme dévie la lumière, mais si vous réalisez un jour un TP vous verrez que pour obtenir une image correcte à travers un prisme, il faut travailler dans une position particulière du prisme correspondant au minimum de déviation, en réalité le prisme n'est un système approximativement stigmatique que s'il est utilisé au minimum de Déviation, il est donc temps de troyver comment placer le prisme pour être au minimum de déviation... Commençons par la relation \ref{relation4}, qui permet d'écrire :













Indice optique d'un prisme
L'indice optique d'un prisme caractérise souvent la manière avec laquelle les rayons seront déviés lors de la traversée de celui-ci. C'est aussi l'indice optique qui limitera l'utilisation du prisme dans certains instruments tels que les réfractomètres, il est, vous l'aurez compris, très important de savoir déterminer l'indice optique d'un prisme, et la déviation minimale pourra nous aider dans cette tache! La relation \ref{relation4} combinée aux relations obtenues sur la déviation minimale permet d'écrire :


jusqu'ici, nous avons décrit le prisme, étudier les relations que nous fournissent l'optique géolétrique et nous avons donc expliquer la déviation du faisceau lumineux par un prisme mais nous n'avons en aucun cas expliqué la décomposition de la lumière blanche or tel êtait notre objectif, il serait donc temps de s'y atteler...
Dispersion du prisme
Puisque les relations géométriques n'expliquent pas l'expérience de Newton, il faut rajouter de l'optique ondulatoire, et en l'occurence une dépendance de l'indice optique (encore lui!) avec la longueur d'onde. En effet l'indice optique d'un milieu est fonction de la longueur d'onde, la relation porte le nom de relation de Cauchy :


 ). Mais soyons un peu moins ambitieux et commençons par étudier la variation de la déviation D avec l'indice optique, notre objectif étant d'étudier la fonction D(n) il nous faut exprimer la dérivée
). Mais soyons un peu moins ambitieux et commençons par étudier la variation de la déviation D avec l'indice optique, notre objectif étant d'étudier la fonction D(n) il nous faut exprimer la dérivée  . Pour cela, reprenons la relation donnant l'indice du prisme et différencions-la :
. Pour cela, reprenons la relation donnant l'indice du prisme et différencions-la :
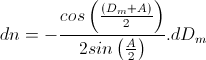

Dm=79,17
Et on en déduit grace à la formule de la déviation minimale (Calcul en degré) :
 ° Ce n'est pas énorme, et pour observer la dispersion d'un prisme, ilfaudra donc de la place et donc des montages encombrants! D'autre part, ce qui fait la dispersion du prisme fait aussi ses défauts: l'interaction matièe rayonnement qui est fonction de la longueur d'onde et de l'indice optique... cette interaction est aussi respondable d'absorption et le prisme qui est transparent pour la plupart du visible devient parfaiteent opaque aux UV ce qui limite son domaine d'utilisation, il a donc fallu trouver plus efficace....
° Ce n'est pas énorme, et pour observer la dispersion d'un prisme, ilfaudra donc de la place et donc des montages encombrants! D'autre part, ce qui fait la dispersion du prisme fait aussi ses défauts: l'interaction matièe rayonnement qui est fonction de la longueur d'onde et de l'indice optique... cette interaction est aussi respondable d'absorption et le prisme qui est transparent pour la plupart du visible devient parfaiteent opaque aux UV ce qui limite son domaine d'utilisation, il a donc fallu trouver plus efficace....
III. Le réseau
1. Description
Imaginé par un certain Fraunhofer le réseau est un ensemble de fentes très fines placées périodiquement les unes à coté des autres, la distance entre deux fentes est appelée \emph{pas} du réseau (souvent noté a)et est de l'ordre du micromètre. Le plus souvent ce n'est pas le pas du réseau qui est indiqué par les fabricants mais le nombre de traits par millimètre, n, la relation liant ces deux grandeurs n'étant autre que :

2. Formule du réseau
Pour expliquer la dispersion du réseau, considérons deux fentes fines consécutives éclairées par un faisceau de lumière parallèle arrivant avecune incidence i sur le réseau. Les fentes étant chacune très fine, on observera diffraction du rayon lumineux et des rayons vont émerger de l'autre cotê avec des angles quelconques mais nous nous intéresserons à des rayons qui émergent du réseau avec un angle que nous noterons i'. Il existe entre les deux rayons émergents une différence de marche qu'il nous faut calculer.La situation est représentée par la figure suivante.
qu'il nous faut calculer.La situation est représentée par la figure suivante.



s'obtient en utilisant la convention de signe décrite sur la figure. On retiendra donc :




La plupart du temps les réseaux sont utilisés en incidence normale, la formule fondamentale devient alors :

- La position des maxima d'intensité dépend de la longueur d'onde : un réseau est donc un système dispersif et l'origine de la dispersion est le phénomène d'interférence (certains auteurs diront diffraction)
- Quelque soit la longueur d'onde, pour k=0 il y a un maximum, on observera donc un mélange de toute les logueurs d'onde au centre.
- pour k=1, les angles theta sont souvent faibles et on peut se permettre l'approximation
 et la dispersion pourra alors être considérée comme linéaire.
et la dispersion pourra alors être considérée comme linéaire.
3. Dispersion
La dispersion d'un réseau est définie par :

- Plus le pas a du réseau est petit, plus la dispersion est grande, en conséquence plus le nombre de traits par millimètre est grand, plus la dispersion du réseau est bonne
- Plus l'ordre k d'interférence est grand, plus la dispersion est grande. A priori il faudrait donc travailler avec un ordre d'interférence grand, mais cela se fait très peu en pratique car d'une part la luminosité diminue avec l'ordre (pour la plupart des réseaux, k ne voit plus de raies après l'ordre 3) et d'autre part il y a chevauchement des ordres. Pour travailler dans un ordre élever en s'affranchissant de ces inconvénients, on utilise des réseaux blazés utilisés en réflexion et dans lesquels les fentes sont remplacés par des miroirs légèrement inclinés. On n'observe plus d'ordre 0 et on concentre toute l'énergie lumineuse (ou du moins une grande partie) dans un ordre élevé afin d'augmenter la dispersion du réseau.
 soit assimilé à 1. On obtient alors
soit assimilé à 1. On obtient alors

 soit environ 12°, c'est mieux que pour le prisme et c'est ce qui justifie majoritairement l'emploi du réseau même si on peut faire encore mieux...\\
La suite au prochain épisode!
soit environ 12°, c'est mieux que pour le prisme et c'est ce qui justifie majoritairement l'emploi du réseau même si on peut faire encore mieux...\\
La suite au prochain épisode!
I Le spin
I.1 Brève introduction à la physique quantique
Pour commencer, je tenais à vous donner un bref aperçu de l’immensité du travail et des réflexions qui ont
permis l’émergence de toutes les choses que nous allons voir dans ce chapitre. En effet, tous les phénomènes
dont nous allons maintenant parler n’ont qu’une seule origine : la physique quantique ; ils proviennent donc
d’un monde qui ne ressemble en rien à ce que vous connaissez, Feynman a écrit : ≪ Le comportement de la
matière à petite échelle est différent tout simplement. Un atome ne se comporte pas comme un poids qui
oscille au bout d’un ressort. Il ne se comporte pas non plus comme un modèle réduit du système solaire […]. Il
ne ressemble à rien que vous ayez déjà vu. Il y a quand même une simplification. Les électrons, de ce point
de vue, se comportent exactement comme les photons ; ils sont tous loufoques, mais exactement de la même
façon ≫.
Cette citation va prendre tout son sens dans les paragraphes qui suivent, ce qu’il faut surtout en retenir,
c’est que la nature est ce qu’elle est, parfois il ne faut pas chercher à la comprendre, mais se limiter à la
décrire. Vers 1925, la description de la nature a mené les physiciens à élaborer une théorie qui s’avéra
bien plus grande que ce qu’ils avaient imaginé, cette Physique Quantique qui vit alors le jour
est d’une simplicité étonnante pour quiconque accepte d’abandonner toute intuition et sens
≪ logique ≫ car la logique de la nature est parfois trop complexe pour nous. Ce cours en est une
démonstration étonnante, la résonance magnétique nucléaire est l’une des techniques d’analyse des plus
performantes et des plus utilisées, et pourtant son origine n’a pas d’équivalent classique : Le
spin.
C’est donc sur cette étrange caractéristique des objets atomiques et subatomiques, sur ce spin, que nous allons travailler durant ces quelques heures. Le formalisme mathématique de la physique quantique vous étant parfaitement étranger, nous nous limiterons à une description et à l’énoncé des principaux résultats.
I.2 L’expérience de Stern et Gerlach
Le spin dont je vous parlais quelques lignes plus haut n’est pas une fantaisie de physiciens, pour le prouver,
intéressons nous à une expérience historique : l’expérience de Stern et Gerlach.
Un faisceau monocinétique d’atome d’argent propulsé dans une zone où règne un champ magnétique perpendiculaire à la direction du faisceau, soit Oz l’axe portant ce champ. Les atomes étant neutres, la force de Lorentz est nulle, de ce fait on s’attend à ce que les atomes continuent leur course suivant une trajectoire rectiligne uniforme. Le résultat de l’expérience est tout autre, on observe la séparation du faisceau en deux faisceaux de même taille, l’un des faisceaux est dévié vers le haut, l’autre est dévié vers le bas. Comment cela est-il possible ? Si la trajectoire d’une particule n’est pas rectiligne uniforme c’est que cette particule est soumise à une force, l’origine de cette force ne peut être due qu’à la présence d’un moment magnétique propre à la particule (la force de Lorentz étant nulle). En effet lorsqu’une particule possède un moment magnétique, son énergie potentielle en présence d’un champ magnétique est donnée par :

Supposons que le champ magnétique soit nul suivant les directions Ox et Oy, il est donc dirigé suivant l’axe
 et de surcroît, il n’est pas constant dans cette direction. On peut alors montrer qu’il existe une force dont
l’expression est :
et de surcroît, il n’est pas constant dans cette direction. On peut alors montrer qu’il existe une force dont
l’expression est :

C’est cette force qui est à l’origine de la déviation du faisceau. Nous venons d’expliquer pourquoi le
faisceau est dévié, mais jusque là le champ magnétique est inhomogène certes, mais il peut
prendre un ensemble continu de valeurs, il décroit de manière continue lorsqu’on s’éloigne du
dispositif. Par conséquent, on s’attend à observer une grosse tache sur l’écran et non pas deux
taches bien distinctes. Pour interpréter ces deux taches, une seule hypothèse est plausible : le
moment magnétique est quantifié, et dans le cas des atomes d’argent, il ne peut prendre que deux
valeurs.
On montre en physique quantique que le moment magnétique d’une particule est relié au moment cinétique de celle-ci par :
 | (17.1) |
Où :
- g est le rapport gyromagnétique
- Jz est le moment cinétique de la particule suivant l’axe Oz
- e la charge élémentaire
- m la masse de la particule
Mais qu’est ce donc que cette chose étrange : le moment cinétique ? Pour ceux d’entre vous qui auraient déjà entendu parler de rotation d’un solide ce terme doit évoquer quelque chose, en effet un moment cinétique est caractéristique (en mécanique classique) de la rotation, c’est en quelque sorte l’équivalent de la vitesse de translation pour les mouvements de translation. Peu importe, cette notion est classique, laissons là donc de coté et restons dans le monde quantique où il existe deux types de moments cinétiques :
- Un moment cinétique orbital, L, auquel sont associés un rapport gyromagnétique de 1 et le nombre quantique l. Ce nombre quantique ne peut prendre que des valeurs entières, en particulier, on peut avoir l = 0. Or si l = 0, ml = 0 et le faisceau n’est pas dévié comme on le constate expérimentalement : le moment cinétique orbital ne peut pas expliquer l’expérience de Stern.
- Un moment cinétique, S, auquel on associe un nombre quantique s pouvant prendre des valeurs
entières et demi-entières, en particulier on peut avoir S =
 . La projection de ce moment
cinétique sur l’axe Oz ne peut alors prendre que deux valeurs ms = ±
. La projection de ce moment
cinétique sur l’axe Oz ne peut alors prendre que deux valeurs ms = ± à chacune de ces valeurs
correspond une tache : l’expérience de Stern est expliquée.
à chacune de ces valeurs
correspond une tache : l’expérience de Stern est expliquée.
En conclusion, on retiendra que toute particule possède un moment cinétique intrinsèque appelé spin, la projection de ce moment cinétique sur l’axe Oz ne peut prendre que des valeurs entières ou demi entières.
17.1.3 Le spin 
Nous allons, pour simplifier les choses dans la suite de ce cours, nous intéresser à des particules de spin  ,
peut-être pensez vous qu’il s’agit là d’une restriction un peu trop grande, aussi pour vous en dissuader nous
allons donner quelques exemples de particules de spin
,
peut-être pensez vous qu’il s’agit là d’une restriction un peu trop grande, aussi pour vous en dissuader nous
allons donner quelques exemples de particules de spin  .
.
L’électron
L’électron est pour l’instant une particule élémentaire totalement indivisible, il est caractérisé par la charge
élémentaire e et par un spin s=  . Il lui est associé :
. Il lui est associé :
- Un moment cinétique orbital avec un facteur gyromagnétique de 1
- Un moment cinétique de spin avec un facteur gyromagnétique de 2 (environ)
Les nucléons
Les nucléons ne sont pas, à l’instar de l’électron des particules élémentaires, ils sont constitués de particules encore plus petites : les quarks. On distingue deux catégories de nucléons :
- Les protons qui possèdent un spin
 et un rapport gyromagnétique de 5,5883
et un rapport gyromagnétique de 5,5883
- Les neutrons qui possèdent aussi un spin
 mais avec un rapport gyromagnétique de -3,8263
mais avec un rapport gyromagnétique de -3,8263
Dans l’expérience de Stern et Gerlach, nous avons posé l’hypothèse du spin  sans la justifier, revenons
maintenant sur la justification. L’atome d’argent possède 47 électrons, c’est donc un système étonnamment
compliqué et pourtant, il ne fournit que 2 taches. Eh bien oui cet ensemble complexe se comporte dans
l’expérience de Stern comme un simple électrons, en effet les électrons sont tous appareillés sauf 1, le spin
total des électrons est donc de
sans la justifier, revenons
maintenant sur la justification. L’atome d’argent possède 47 électrons, c’est donc un système étonnamment
compliqué et pourtant, il ne fournit que 2 taches. Eh bien oui cet ensemble complexe se comporte dans
l’expérience de Stern comme un simple électrons, en effet les électrons sont tous appareillés sauf 1, le spin
total des électrons est donc de  . Reste un dernier point à éclaircir, les nucléons ont aussi un spin,
comment cela se fait-il qu’ils n’interviennent pas dans l’expérience de Stern ? La réponse vient de
l’expression 17.1, le moment magnétique d’une particule est inversement proportionnelle à sa
masse, la masse d’un proton étant environ 1836 fois plus forte que celle d’un électron, le moment
magnétique du proton est beaucoup plus faible que celui de l’électron (657 fois). Un champ
magnétique réglé pour séparer les projections du moment magnétique de l’électron ne permet pas une
séparation visible des composantes du moment magnétique du proton (ou du neutron), voilà
pourquoi l’expérience de Stern peut s’expliquer à partir du seul spin d’un électron : le spin
. Reste un dernier point à éclaircir, les nucléons ont aussi un spin,
comment cela se fait-il qu’ils n’interviennent pas dans l’expérience de Stern ? La réponse vient de
l’expression 17.1, le moment magnétique d’une particule est inversement proportionnelle à sa
masse, la masse d’un proton étant environ 1836 fois plus forte que celle d’un électron, le moment
magnétique du proton est beaucoup plus faible que celui de l’électron (657 fois). Un champ
magnétique réglé pour séparer les projections du moment magnétique de l’électron ne permet pas une
séparation visible des composantes du moment magnétique du proton (ou du neutron), voilà
pourquoi l’expérience de Stern peut s’expliquer à partir du seul spin d’un électron : le spin  .
.
II Spin dans un champ magnétique constant
II.1 Interaction spin champ magnétique
On considère une particule de spin  occupant une position fixe dans l’espace et on plonge cette particule
dans un champ magnétique constant dirigé suivant l’axe Oz. On se propose d’étudier l’évolution du spin
provoquée par le champ magnétique au cours du temps. Pour décrire la situation, il faut reprendre les
éléments qui nous ont permis d’interpréter l’expérience de Stern et les appliquer un à un à la
situation du spin
occupant une position fixe dans l’espace et on plonge cette particule
dans un champ magnétique constant dirigé suivant l’axe Oz. On se propose d’étudier l’évolution du spin
provoquée par le champ magnétique au cours du temps. Pour décrire la situation, il faut reprendre les
éléments qui nous ont permis d’interpréter l’expérience de Stern et les appliquer un à un à la
situation du spin  . A toute particule de spin non nul est associé un moment magnétique
. A toute particule de spin non nul est associé un moment magnétique  défini
par :
défini
par :
 | (17.2) |
Lorsque ce moment magnétique est placé dans un champ magnétique , il est soumis, selon la mécanique classique, à un couple :
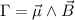 | (17.3) |
Il résulte de cette interaction une énergie potentielle :
 | (17.4) |
Ces trois équations traduisent l’interaction spin-champ magnétique, voyons maintenant les conséquences physiques de chacune d’entre elle.
II.2 Précession de Larmor
En mécanique classique, lorsqu’on applique un couple sur un système, le système évolue de sorte à annuler
ce couple et retrouver l’équilibre. La situation est quelque peu différente en quantique (comme toujours !). Le
spin  soumis à un champ constant tend effectivement à s’aligner avec le champ mais n’y parvient pas
complètement à cause de la quantification de la composante sz. On montre que les composantes du moment
cinétique de spin sont régies par les équations :
soumis à un champ constant tend effectivement à s’aligner avec le champ mais n’y parvient pas
complètement à cause de la quantification de la composante sz. On montre que les composantes du moment
cinétique de spin sont régies par les équations :

Où on a noté la pulsation de Larmor :

Les équations précédentes décrivent un mouvement dit de précession, le moment magnétique tourne autour de l’axe Oz comme une toupie , ce mouvement se fait à la pulsation de Larmor.

II.3 Champ magnétique et niveaux énergétiques
Utilisons les relations 17.2 et 17.4 pour exprimer de manière un peu plus précise l’énergie potentielle
d’interaction entre un champ magnétique et un spin  , on obtient :
, on obtient :

Où nous avons désigné par sz la projection sur l’axe Oz du spin. Pour un spin  cette projection ne peut
prendre que deux valeurs déterminée sz = +
cette projection ne peut
prendre que deux valeurs déterminée sz = + et sz = -
et sz = - . En conséquence, il n’existe que deux états
d’énergie possible pour le système :
. En conséquence, il n’existe que deux états
d’énergie possible pour le système :


Nous avons là une conséquence fondamentale de l’interaction spin-champ, le champ magnétique provoque la séparation des niveaux énergétiques en deux sous-niveaux comme illustré par la figure 17.2.

La particule a donc le choix entre deux niveaux énergétiques, en conséquence on trouvera des particules sur le niveau du bas et des particules sur le niveau du haut, les particules étant généralement partisantes du moindre effort, il y aura plus de particules sur le niveau du bas que sur le niveau du haut. Le rapport des populations est donné par la répartition de Maxwell-Boltzmann.

On notera que l’écart des niveaux d’énergie est fonction du champ magnétique B0  , pour bien
discerner les niveaux, il faut donc appliquer un champ magnétique intense qui soit au moins de l’ordre du
Tesla.
, pour bien
discerner les niveaux, il faut donc appliquer un champ magnétique intense qui soit au moins de l’ordre du
Tesla.
III Spin et champ oscillant : La résonance magnétique nucléaire
III.1 Magnétisme nucléaire
Otto Stern reçoit en 1946 le prix Nobel pour l’expérience réalisée en collaboration avec Gerlach mais il ne
s’était pas arrêté là et poursuivit ses investigations. Quelques temps plus tard il réussit à mettre en évidence
et à mesurer le moment magnétique du proton. Cette découverte est lourde en conséquence : il existe un
magnétisme nucléaire !
Vous vous rappelez du magnétisme (??) : les substances ferromagnétiques, diamagnétiques et paramagnétiques, et bien tout cela existe aussi au niveau nucléaire, ce problème intéresse beaucoup de monde y compris un certain Rabi. Né en 1899, il a fait une thèse sur le magnétisme à Cornell et arrive à Columbia, son expérience du magnétisme lui permet d’aller encore plus loin que Stern et il obtient une précision 1000 fois supérieure à celle de Stern sur la mesure du moment magnétique nucléaire. C’est grâce à cette précision extraordinaire que Rabbi va mettre au point la Résonance magnétique nucléaire.
III.2 Champ tournant
La méthode de Rabi est tout simplement géniale, il propose de superposer à un champ fixe constant,  , un
champ tournant
, un
champ tournant  . Détaillons quelque peu la constitution de ce champ tournant. Comment peut-on
créer un champ magnétique décrivant un cercle ? Nous allons nous aider d’un résultat que nous
avons légèrement abordé lors de notre étude de la polarisation : la composition de deux vecteurs
rectangulaires oscillants déphasés de
. Détaillons quelque peu la constitution de ce champ tournant. Comment peut-on
créer un champ magnétique décrivant un cercle ? Nous allons nous aider d’un résultat que nous
avons légèrement abordé lors de notre étude de la polarisation : la composition de deux vecteurs
rectangulaires oscillants déphasés de  a pour résultat un vecteur décrivant un cercle au cours du
temps.
a pour résultat un vecteur décrivant un cercle au cours du
temps.
En conséquence, pour créer un champ tournant, on va créer deux champ magnétiques oscillants l’un suivant Ox l’autre suivant Oy et ça c’est quelque chose que nous savons faire : Le champ magnétique dans un solénoïde est porté par l’axe du solénoïde et est proportionnel au courant I qui parcourt ses spires, le champ total que nous devons créer est donc la composition de trois champ comme indiqué sur la figure 17.4.

III.3 Résonance Magnétique
Ce champ magnétique tournant va provoquer des changements remarquables sur l’état de spin. Pour le voir, nous allons suivre les traces de Rabi. Il réalise en 1939 l’expérience décrite par la figure 17.5.

Il s’agit de deux appareils de Stern et Gerlach entre lesquels, on a introduit un dispositif de champ tournant et fixe que nous avons décrit plus haut. Tout d’abord en l’absence de champ tournant, les particules dans l’état d’énergie le plus bas (c’est-à-dire celles ayant un spin dirigé vers le haut) sont déviées par le premier le premier appareil vers le haut et sont déviés vers le bas par le second : elles atteignent donc le détecteur. Introduisons maintenant le champ tournant, on constate alors que le signal capté par le détecteur dépend fortement de la fréquence du champ tournant, et passe par un minimum bien marqué pour .
L’introduction d’un champ magnétique tournant provoque pour ω = ωL le basculement du spin qui passe alors dans l’état d’énergie supérieur : il y a résonance. La fréquence de résonance est déterminée, d’une part par l’intensité du champ B0 mais aussi et par les caractéristiques de la particule (rapport gyromagnétique et masse).
IV Principe de la spectroscopie RMN
IV.1 Niveaux d’énergie et noyaux
La résonance magnétique nucléaire n’est rien d’autre que l’application de l’expérience de Rabi au spin nucléaire. Pour cela, il faut donc que le spin ne soit pas nul. Pour un noyau de Z protons et (Z-A) neutrons le spin sera :
- Demi entier si A et Z sont impairs
- Nul si A et Z sont Pairs
- Entier si A est pair et Z impair
Le noyau le plus utilisé en spectroscopie est le proton 11H. Le spin du proton est un spin  le problème est donc
parfaitement bien décrit par ce que nous venons de faire. Jusqu’ici nous avons travaillé en phase
gazeuse, il s’agissait de jet moléculaire, or l’intérêt est d’appliquer cette méthode aux phases
condensée. Un problème survient alors, le rapport des populations est donné par le facteur de
Boltzmann :
le problème est donc
parfaitement bien décrit par ce que nous venons de faire. Jusqu’ici nous avons travaillé en phase
gazeuse, il s’agissait de jet moléculaire, or l’intérêt est d’appliquer cette méthode aux phases
condensée. Un problème survient alors, le rapport des populations est donné par le facteur de
Boltzmann :

Pour obtenir la meilleure sensibilité possible, on doit favoriser l’état de plus basse énergie, deux solutions s’offrent alors à nous qui sont toutes deux employées :
- Une diminution de la température
- Une augmentation du champ B0
En combinant ces deux conditions, on obtient des spectres de plus en plus précis qui révéleront la structure fine de la matière.
IV.2 Le déplacement chimique
C’est ici que nous allons rassembler toutes nos connaissances sur la résonance magnétique nucléaire :
- Un noyau, caractérisé par un spin non nul, placé dans un champ magnétique constant possède un moment magnétique oscillant autour de ce champ à la pulsation ωL qui dépend de ce noyau et surtout du champ magnétique
- Il existe au niveau nucléaire un magnétisme (paramagnétisme, diamagnétisme)
- On peut mesurer exactement la fréquence de résonance d’un noyau en utilisant un champ tournant
A ces notions, nous allons rajouter l’environnement chimique du noyau. Il est entouré d’électron plus ou moins
liés en fonction des atomes qui l’entourent. Ces électrons possèdent un spin, et par conséquent possèdent un
moment magnétique.
L’environnement chimique d’un noyau agit comme une multitude de petits aimants (les moments magnétiques) qui modifient le champ magnétique réellement perçu par le noyau. On appelle champ efficace le champ perçu par le noyau et on rassemble sous l’appellation constante d’écran σ l’effet de l’environnement chimique, on a alors :

La constante d’écran comprend trois contributions :
- Une contribution diamagnétique
- Une contribution paramagnétique
- Une contribution due au solvant dont on peut s’affranchir en réalisant la mesure à diverses concentrations
Le terme prédominant reste le terme diamagnétique.
La conséquence de tout ceci est remarquable : en mesurant la fréquence de résonance d’un noyau, on peut retrouver son environnement chimique. En effet l’énergie séparant les niveaux d’un proton libre est plus grande que celle séparant les niveaux d’un proton lié. Pour quantifier tout ceci, on doit introduire une référence, c’est-à-dire un composé inerte pour lequel la fréquence de résonance du proton est fixe. La référence est le tétraméthylsilane. On définit alors le déplacement chimique par :

Une mesure de spectroscopie RMN consiste donc à mesurer la fréquence de résonance des protons constituant un composé et à la comparer à la fréquence de résonance du tétraméthylsilane, le déplacement chimique résultant de cette comparaison est caractéristique de l’environnement du proton.
IV.3 Remarques
Le principe d’une mesure RMN que nous venons d’énoncer est à compléter par quelques éléments :
- La structure fine. Intéressons nous au cas de l’éthanol par exemple, à basse résolution, on obtient trois
signaux dans les rapports 3 :2 :1. Si on augmente la valeur du champ B0 pour augmenter le nombre de
signaux augmente : c’est la structure fine du spectre que nous avons représenté figure 17.6.
La structure fine est le résultat de l’interaction spin-spin c’est-à-dire de l’interaction entre les moments magnétiques des protons.
- Nous avons évoqué les niveaux d’énergie et leur population. Nous avons pourtant soigneusement évité une question, puisque lors de la résonance, les protons acquièrent l’état d’énergie le plus élevé, il doit arriver un moment où ils sont tous dans cet état d’énergie et le phénomène s’arrête, non ? En réalité, les noyaux retrouvent leur état stable en interagissant avec le milieu extérieur : c’est le phénomène de relaxation. Il existe deux types de relaxation possédant des temps caractéristiques différents :
- La relaxation spin-réseau (longitudinale) où le spin échange de l’énergie avec le milieu (solvant, cristal..) la population du niveau supérieur diminue alors et le temps de relaxation T1 est de l’ordre de 0,3 à3s
- La relaxation spin-spin (transversale) qui se fait quasiment sans échange d’énergie et qui ne fait pas
varier la population de niveaux. Le temps caractéristique (T2) de ce phénomène est de 30 à 150ms


V Les spectromètres RMN
V.1 Les premiers spectromètres RMN
Les premiers spectromètres RMN fonctionnent suivant le principe du dispositif de Rabi, la fréquence du champ est maintenue constante et on faisait varier le champ magnétique B0. On repérait ainsi la résonance à l’aide du champ.
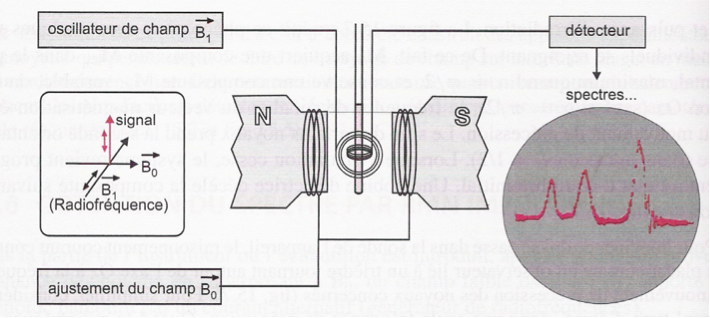
Cette méthode de spectroscopie par absorption a presque totalement été abandonnée aujourd’hui au profit d’une RMN impulsionelle.
V.2 Les spectromètres modernes
En pratique, on applique un champ fixe intense B0 et on lui superpose un champ B1 perpendiculaire à B0,
en effet on montre que les résultats obtenus pour un champ tournant sont équivalents à ceux
obtenu pour un seul champ oscillant (la justification se trouve dans la discussion de II.2). Le
détecteur est un bobinage placé perpendiculairement à B0 et B1. Avant l’application du champ B1
tous les moments magnétiques sont dirigés vers le haut et tournent à des phases différentes, en
moyenne, le moment magnétique résultant est dirigé suivant Oz. Lorsqu’on applique le champ B1 les
moments magnétiques s’accordent entre eux et acquièrent une phase commune, il résulte une
composante non nulle du moment magnétique dans le plan xOy, c’est cette composante que l’on
mesure.
L’appareil envoi une impulsion, c’est-à-dire un signal très court dans le temps mais très étalé en fréquence,
toutes ces fréquences sont ressenties par l’échantillon et le phénomène de résonance magnétique nucléaire se
déclenche pour chaque noyau. Pendant le retour à l’équilibre des noyaux, l’appareil enregistre un signal qui
s’amorti au cours du temps, ce signal est appelé signal de décroissance d’induction libre (FID). L’allure de ce
signal est donnée sur la figure 17.9.

Le traitement de ce signal par transformée de Fourier mène à un spectre classique de RMN. Ce type d’appareil correspond à une méthode simultanée.
1 La fluorescence X
1.1 L’émission de rayons X dans les atomes
Dans ce chapitre nous allons essentiellement parler de rayons X, ce rayonnement tire son nom de
l’incertitude qui régnait autour de sa nature lors de sa découverte. Aussi pour que l’ignorance de ses
découvreurs ne soit pas votre ignorance, rappelons que les rayons X ne sont autres qu’une onde
électromagnétique (comme la lumière visible), le domaine des rayons X correspond à des longueurs
d’ondes allant de 10nm à 1pm. Il s’agit donc d’un rayonnement très énergétique puisque les
photons associés à un tel rayonnement ont une énergie E = hνdont l’énergie peut aller jusque
1,2MeV.
En comparant cette énergie à l’énergie des photons responsables des transitions dans le visible on constate
que la différence est phénoménale, ainsi il semblerait que les transitions électroniques ne peuvent
en aucun cas être responsable de l’émission de rayons X. Ce constat n’est que partiellement
vrai, il est, en effet, vrai que les niveaux électroniques que nous avons considérés jusqu’ici ne
sont absolument pas concernés par l’émission de rayons X en revanche, l’émission de rayons
X par un atome a pour cause le déplacement d’un électron d’un niveau d’énergie à un autre.
Imaginons que pour une raison ou pour une autre, un atome perde un électron de cœur, un électron de la
couche K par exemple. Cet atome ionisé est particulièrement instable, aussi pour combler cette lacune, un
électron d’une couche supérieure doit quitter son rang pour venir combler la lacune existante sur la couche
K, c’est lors de ce passage qu’il y a émission de rayons X, la longueur d’onde émise dépend de la différence
d’énergie entre les niveaux concernés.
La figure 1 montre les différentes possibilités de transition ainsi que la nomenclature des transitions :
- La transition porte le nome de la couche vers laquelle se dirige l’électron, par exemple si une lacune a été créée sur la couche K, on assistera à une transition K…
- En indice on note en lettre grecque minuscule la provenance de l’électron, si l’électron provient de la première couche la plus proche ce sera la transition Kα, si l’électron provient de la deuxième couche juste supérieure, ce sera la transition Kβ etc.
Pour finir on voit qu’il existe une transition limite correspondant à la chute d’un électron non lié (extérieur à l’atome) sur la couche considérée. Nous clôturerons ce paragraphe par un petit exemple. Considérons un noyau de cobalt pour lequel l’énergie de liaison d’un électron de la couche K est EK = 7,71.103eV et c elle d’un électron de la couche L est EL = 7,65.102eV . Si on observe la transition Kα elle sera caractérisée par une raie de longueur d’onde caractérisée par :

Soit λ = 0,179nm
1.2 Condition de fluorescence X
Il existe en réalité deux conditions de fluorescence X : la première porte sur l’ionisation de l’atome
et la seconde sur l’atome lui-même. Le paragraphe précédent montre qu’il y a émission d’un
photon X lorsqu’un électron vient combler une lacune électronique créée sur une couche interne
du noyau. En conséquence pour les atomes ne possédant aucun électron sur la couche L, la
fluorescence X est impossible : même si il se crée une lacune électronique sur la couche K, il n’y a
pas d’électron disponible pour réaliser la transition Kα, et par conséquent pas de rayonnement
X.
Intéressons nous maintenant à la condition d’ionisation de l’atome. Les électrons des couches internes sont
les électrons les plus liés à l’atome, par conséquent les arracher nécessite beaucoup d’énergie.
Deux méthodes sont couramment employées pour arracher les électrons des couches internes et
nous allons les étudier toutes deux. La première méthode consiste à envoyer sur l’atome un
faisceau d’électron monocinétique (cf. cours sur les sources lumineuses). Soit Ei l’énergie de liaison
d’un électron sur la couche cette énergie de liaison correspond à l’énergie qu’il faut fournir pour
arracher l’électron de son orbite et l’extraire de l’atome. La condition d’ionisation de l’atome s’écrit
donc :
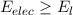
Or l’énergie de l’électron incident, c’est son énergie cinétique dont l’expression est selon le paragraphe ?? :

Où U est la différence de potentiel sous laquelle a été accéléré l’électron.
La seconde méthode consiste à envoyer sur l’atome un faisceau de rayons X. Les photons constituants ce
faisceau possèdent l’énergie :

Pour ioniser l’atome, l’énergie d’un photon doit être supérieure ou égale à l’énergie de liaison de l’électron considéré ( cg. paragraphe ??), ce qui s’écrit :
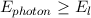
Bien souvent, on utilise un faisceau d’électron pour produire des rayons X dans les tubes sources (Coolidge, Crompton…), on obtient alors un faisceau de rayons X dit primaires. Ces rayons X primaires sont ensuite envoyés sur l’échantillon à étudier et provoque l’émission de rayons X secondaires : c’est la fluorescence X. Finalement ce sont ces rayons X secondaires qui vont être exploités pour déduire les propriétés de l’échantillon.
1.3 Allure des spectres
Les spectres de rayons X sont composés de raies fines, la longueur d’onde d’émission de ces raies est caractéristique de l’atome et dépend très peu de l’environnement chimique de celui-ci. Ainsi la fluorescence X est principalement utilisée pour déterminer les constituants d’un mélange ou déceler la présence de certains atomes dans une substance. D’après ce que nous avons dit précédemment, toutes transition correspondant au passage d’une couche supérieure vers une couche interne provoque l’émission de photon X, on s’attend donc à observer une multitude de raies ce qui serait difficilement exploitable. La nature étant bien faite, le problème se simplifie grandement par l’existence de règles de sélection. Il existe deux règles de sélection qui concerne les nombres quantique orbital l et le moment cinétique j :

Ces conditions limitent fortement le nombre de transitions possibles si bien qu’on peut les représenter sur le schéma de la figure 2.

2 Eléments constitutifs des spectromètres
2.1 Les sources
Les sources de rayons X utilisées dans les spectrophotomètres de laboratoire ont été décrites dans le paragraphe??, on rappelle seulement ici leur nom :
- tube de Coolidge
- tube de Crookes
- tube à anode tournante
Chacun de ces tubes utilise un faisceau d’électrons fortement accélérés par une différence de potentiel pour produire les rayons X, ils sont par conséquent consommateur d’énergie et peu adaptés pour des appareils de mesure in situ. Pour ce type d’appareil in situ ou portatifs, la source est le plus souvent une source radioactive, en effet la capture électronique (cf. ) par exemple est un mode de désintégration radioactive aboutissant à une lacune électronique sur une couche interne, et par suite à l’émission de rayons X. L’inconvénient de ce type de source est qu’il faut détenir une autorisation pour posséder une source radioactive et d’autre part le stockage d’une source radioactive est une affaire délicate, aussi ces sources sont évitées à chaque fois que faire se peut.
2.2 Système dispersif
Tout comme dans le spectromètre UV/Visible les spectrophotomètres de Rayons X doivent disposer d’un
système dispersif. Pourquoi ne pas utiliser un réseau me direz vous. La réponse réside en le principe de
fonctionnement de ceux-ci. Un réseau est un système dispersif qui doit son pouvoir de dispersion au
phénomène de diffraction, or l’étude de la diffraction par une fente nous a montré que le phénomène de
diffraction est de plus en plus efficace que l’ouverture est petite devant la longueur d’onde de
la lumière utilisée. Et nous tenons là toute la problématique. Pour que le réseau soit efficace
dans le domaine des rayons X, il faudrait que le pas du réseau soit petit devant la longueur
d’onde X or ceci est difficilement réalisable, en conséquence nous devons trouver un autre système
dispersif.
La solution au problème a été donnée par la cristallographie, cette banche des sciences physiques a permis
d’évaluer la distance entre les atomes d’un réseau cristallin, cette distance est de l’ordre de l’Angström. Or la
longueur d’onde moyenne des rayons X se situe autour de l’Angstrom, pourquoi donc n’utiliserions nous pas
un cristal comme système dispersif.

Un cristal est une structure périodique, tout comme un réseau, il réalise une modulation périodique de
l’amplitude de l’onde lumineuse, étudions donc ce système.
On considère un faisceau lumineux monochromatique arrivant sur le cristal sous une incidence i. ce faisceau émerge du cristal avec un angle d’émergence θ. La situation est tout à fait analogue à celle du réseau et on peut calculer la différence de marche entre les deux rayons rouge et bleu. Lorsque le rayon rouge atteint la surface du cristal, le rayon bleu ne l’a pas encore atteint, il possède un retard AB comme indiqué figure 4.

La longueur AB est AB = asini.
De même à la sortie du cristal, le rayon bleu repart avant le rayon rouge et il existe une différence de marche CD comme indiqué figure 5.

La longueur CD est CD = asinθ.
On introduit maintenant le sens d’orientation des angles : l’angle est compté positivement dans le sens trigonométrique et négativement dans le sens horaire. Finalement la différence de marche s’écrit :

On observera de la lumière (interférences constructives) si :

On retrouve la formule fondamentale des réseaux établie cette fois-ci en réflexion. Dans le cas d’un cristal, on montre que l’énergie lumineuse est réfléchie presque à 100% lorsque l’angle d’incidence est égal à l’angle d’émergence et est nulle ailleurs. De ce fait, la formule devient :
 | (1) |
Cette relation porte le nom de condition de Bragg
Par conséquent un cristal se comporte, pour les rayons X exactement comme un réseau, on peut donc choisir
l’orientation des cristaux de sorte à isoler dans le spectre d’émission des rayons X une longueur d’onde. Nous
retrouverons ce principe lors de l’étude des spectromètres.
2.3 Les capteurs
Les capteurs utilisés dans les spectromètres à rayons X sont des capteurs énergétiques de particules, on compte le nombre de photon X qui arrive ainsi que leur énergie. Il existe deux grands types de capteurs :
- Les capteurs à semi conducteurs, où la détection est assurée par une jonction PN (cf. ??). Pour diminuer le bruit de fond de ces capteurs, on les maintient à basse température par azote liquide.
- Le compteur proportionnel à gaz dans lequel chaque photon X provoque l’ionisation d’un mélange gazeux (argon méthane par exemple) cette ionisation permet le passage du courant et on détecte alors une impulsion proportionnelle à l’énergie du photon.
Dans les deux cas, l’impulsion permet de remonter à l’énergie des photons. Après avoir défini une largeur d’acquisition de 10 à 20 keV, l’analyseur va comptabiliser pendant toute la durée de la mesure les impulsions libérées par les photons reçus. Chaque photon sera classé dans un canal d’énergie correspondant à un intervalle d’énergie de quelques eV. Il en résulte un spectre construit comme un histogramme. On notera que le procédé ci-dessus conduit à une analyse simultanée de tout le spectre (qui sera d’autant meilleure que le temps d’acquisition est grand).
3 Différents types de spectromètres
3.1 Les spectromètres dispersifs en longueur d’onde(WDXRF)
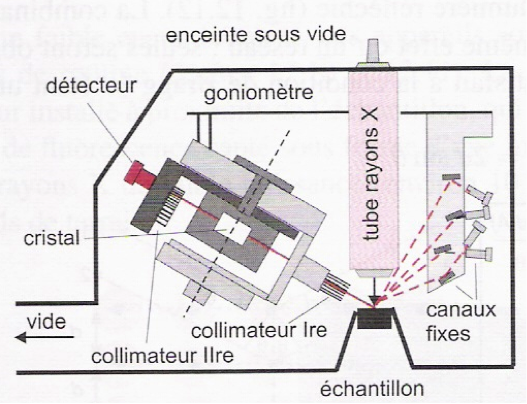
Dans ces appareils connus pour leur très bonne résolution, le rayonnement de fluorescence de l’échantillon traverse un collimateur constitué de longs feuillets métalliques, puis vient frapper un cristal taillé de telle sorte que les atomes constitutifs forment des plans parallèles à la surface. Parmi les analyseurs de ce type, on distingue deux catégories :
- Les analyseurs séquentiels, qui comportent un montage goniométrique permettant la rotation
synchronisée du cristal et du détecteur (cf. figure 6). Ils sont réservés aux dosages des éléments non
routiniers
- Les analyseurs à canaux fixes qui exploitent une conséquence de la loi de Bragg. Cette relation montre
qu’en fixant a (pas du réseau), donc en choisissant convenablement le cristal, et en fixant l’angle de
détection, on isole une longueur d’onde. Partant de ce principe, on peut installer autour de
l’échantillon plusieurs dizaines d’ensemble cristal-détecteur, chaque couple (a, θ) permettant
d’isoler une longueur d’onde, donc de repérer un élément précis avec une grande sensibilité.

3.2 Les spectromètres dispersifs en énergie(EDXRF)
Il s’agit d’appareils de faible encombrement réservés à l’analyse qualitative et aux dosages de routine. Le spectre est obtenu en faisant appel à un détecteur placé à proximité de l’échantillon, qui permet de déterminer l’énergie de chaque photon de fluorescence capté sous forme d’impulsion. Ces appareils sont généralement équipés de sources de faible puissance (10W) ou d’une source radioactive lorsqu’ils sont placés in situ.

4 Applications
4.1 Absorption des rayons X
L’absorption des rayons X par la matière conditionne indirectement l’analyse par fluorescence X.
Nous allons donc modéliser cette absorption par un modèle que nous avons déjà rencontré.
On suppose que l’absorption d’une tranche dx de matière située entre les abscisses x et x + dx est
proportionnelle à l’épaisseur (dx) de cette tranche et au flux lumineux qui arrive sur cette tranche. On
note μ le coefficient de proportionnalité, appelé coefficient linéique d’absorption. On peut alors
écrire :

Soit finalement :

Le coefficient d’absorption linéique est calculable pour tout matériau dont on connaît la masse volumique et la constitution, il existe en effet des tables donnant les coefficients d’absorption massiques des éléments. On a ainsi pu tracer la transmittance du béryllium et de l’aluminium indiquée sur la figure 9.

4.2 Analyse quantitative et autres applications
La relation entre concentration massique en élément et intensité mesurée d’une de ses raies caractéristiques
est complexe. Dans le cas d’analyse de raies, différents modèles ont été développés pour corréler la
fluorescence à la concentration atomique, on doit cependant leur apporter diverses corrections dues aux
interactions interélément, à l’autoabsorbtion, au rendement… Une bonne analyse quantitative est donc chose
délicate à réaliser.
Néanmoins, l’étude des rayons X constitue une excellente méthode d’analyse qualitative, utilisée dans les
industries traitant des métaux, de alliages et d’une façon générale dans la grande industrie minérale
(sidérurgie, industries des ciments, e la céramique, du verre). Cette méthode qui nécessite très peu ou pas de
préparation de l’échantillon a de plus une plage dynamique large, il est possible de doser sur une même prise
d’essai deux éléments dont les concentrations sont de 50% pour l’un et de quelques ppm pour l’autre.
Avec les progrès des détecteurs, les analyses de routines à RX atteignent aujourd’hui la précision des
méthodes d’analyse par voir humide son champ d’application s’est donc notablement étendu.
1 Niveau d’énergie dans les atomes
1.1 Principe de la spectroscopie d’absorption
Les chapitres précédents ont présentés la nature de la lumière, les sources de rayonnements
électromagnétiques et pour finir les capteurs de rayonnement ; toutes ces notions vont être rassemblées ici (et
dans les prochains chapitres) pour construire la spectroscopie des rayonnements électromagnétiques.
Nous commençons notre étude par la spectroscopie UV/ visible. La base de cette méthode très utilisée en
chimie analytique est le constat suivant : toute matière modifie le spectre de la lumière qui la traverse d’une
manière qui lui est propre. L’idée, c’est donc de faire passer au travers la matière un faisceau lumineux et de
comparer le spectre de la lumière avant et après cette traversée. Ce que nous venons d’énoncer n’est autre
que le principe de la spectrométrie d’absorption.
1.2 Les niveaux d’énergie
Intéressons nous à présent à l’interaction matière rayonnement. Les molécules d’un échantillon recevant de la lumière est caractérisée par ses états d’énergie. En effet depuis l’avènement de la mécanique quantique, on sait que les électrons d’un atome, d’une molécule ou même d’un solide sont organisés en niveaux d’énergie quantifiable. D’une manière générale, l’énergie d’une molécule est constituée de trois termes :
- L’énergie électronique
- L’énergie de vibration
- L’énergie de rotation
Ces niveaux d’énergie correspondent à des niveaux dont les écarts sont très différents les uns des autres (cf. figure 1).
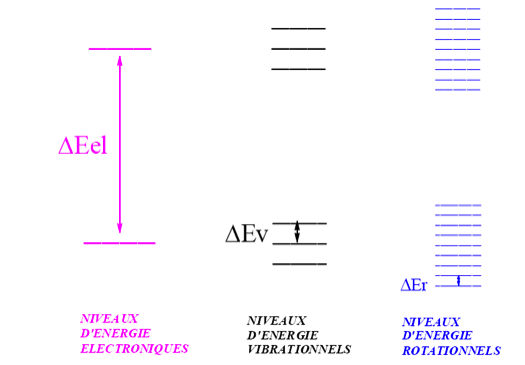
Lorsque la lumière traverse l’échantillon, elle lui fournit de l’énergie ce qui a pour conséquence de faire passer la molécule dans un état excité. Les écarts entre les niveaux d’énergie étant très grands, il en résulte une énergie des radiations nécessaires pour exciter les niveaux électroniques, vibrationnels ou rotationnels est très différentes.
1.3 Domaine spectral et sources
L’énergie d’une radiation est déterminée par sa longueur d’onde. En effet, une radiation lumineuse est constituée de photons dont l’énergie est donnée par E = hν, qu’on pourra aussi écrire :
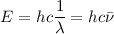
Où ν est le nombre d’onde qu’on exprime souvent en cm-1. Il n’y aura absorption d’un photon que si l’énergie de celui-ci correspond à la différence d’énergie entre deux niveaux :

Commençons par les niveaux d’énergie rotationnels, ceux-ci correspondent tout simplement à différents états de rotation de la molécule. On imagine facilement que passer d’un état de rotation à un autre demande très peu d’énergie, de ce fait pour exciter ces niveaux, il faudra des photons très peu énergétiques. L’excitation des niveaux rotationnels nécessite des photons appartenant au domaine des micro-ondes.

Passons maintenant aux niveaux d’énergie vibrationnels, ceux-ci correspondent à l’énergie nécessaire pour faire entrer en vibration les liaisons de la molécule. On imagine qu’il faut un peu pus d’énergie pour faire vibrer une liaison à la manière d’un ressort que pour la faire tourner sur elle-même, en conséquence il faudra des photons un peu plus énergétiques. L’excitation des niveaux vibrationnels nécessite des photons appartenant au domaine de l’infrarouge.

Finalement reste les niveaux électroniques. Ces niveaux représente l’état énergétique des électrons de la molécules. Le passage d’un niveau à un autre correspond généralement au passage d’un électron d’une orbitale à une autre. Comme exemple, on peut citer le passage de l’électron d’un niveau non-liant à un niveau anti-liant ou d’un niveau liant du système π vers un niveau anti-liant. L’excitation des niveaux électroniques nécessite des photons appartenant au domaine visible ou de l’ultraviolet.

C’est particulièrement à ces dernières transitions que nous allons nous intéresser dans ce chapitre. La figure 2 représente le domaine spectral associé à chaque type de transition.

2 Loi de Beer Lambert
2.1 Historique
La colorimétrie visuelle est l’une des plus anciennes méthodes d’analyse. Déjà appliquée du temps des
grecs et des romains, elle commence à prendre un caractère scientifique avec Pierre Bouguer en
1729, celui dit : ≪ si une certaine épaisseur de verre coloré absorbe la moitié de la lumière issue
de la source, une épaisseur de verre double réduit cette lumière au quart de sa valeur initiale
≫.
Trente ans plus tard, un certain Jean-Henri Lambert reprend cet énoncé et le traduit de manière plus
mathématique : ≪ le logarithme de la diminution de lumière est égal au produit de son opacité par son
épaisseur. ≫
Enfin en 1850, Auguste Beer établit qu’il existe une relation de proportionnalité entre la concentration et
l’absorbance.
2.2 Transmittance, absorbance
Conformément au principe de la spectroscopie d’absorption, on cherche à quantifier l’action d’un milieu sur la lumière qui la traverse. Nous savons qu’un milieu agit sur le spectre de la lumière, autrement dit l’énergie transportée par chaque composante monochromatique est modifiée par la traversée d’un milieu matériel. Pour simplifier le problème, supposons qu’on dispose d’une source parfaitement monochromatique, l’énergie transportée par la radiation qui en résulte est caractérisée par le flux Φ de la source (énergie par unité de temps). Nous devons donc comparer le flux d’un faisceau ayant traversé l’échantillon à analyser et celui d’un faisceau n’ayant pas traversé l’échantillon (faisceau incident). On note :
- Φ0 le flux de la radiation n’ayant pas traversé l’échantillon à analyser
- Φ le flux de la radiation ayant traversé l’échantillon à analyser.
Nous allons voir comment on peut relier ces deux flux en utilisant un modèle simple. On suppose que la traversée d’une épaisseur dx d’échantillon située à l’abscisse x conduit à une absorption (diminution du flux) qui est proportionnelle à la fois à l’épaisseur dx et au flux incident sur cet échantillon i.e Φ(x). (cf. figure 3)

On peut donc écrire :

Ce qui peut aussi s’écrire :

Soit en intégrant entre les abscisses x = 0 et x = l :
 | (1) |
On définit alors la transmittance par :
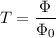 | (2) |
Et l’absorbance par :
 | (3) |
La relation 1 permet alors d’exprimer l’absorbance de l’échantillon :

L’absorbance (parfois appelée densité optique) d’un échantillon est proportionnelle à l’épaisseur d’échantillon traversé, c’est ce qu’avait déjà remarque en 1729 Pierre Bouguer.
2.3 Loi de Beer-Lambert
Nous avons défini l’absorbance au paragraphe précédent et nous avons montrer que l’absorbance A d’un
échantillon est proportionnelle à l’épaisseur d’échantillon traversé : c’est la loi de Lambert.
Cette loi est incomplète, en effet, lorsque que la lumière traverse 10cm d’air ou 10cm de permanganate de
potassium, la modification du spectre ne sera pas la même. Aussi, on se doit d’introduire un coefficient
caractéristique de chaque substance : le coefficient d’extinction molaire, noté ϵ. D’autre part
que la lumière traverse 1cm d’une solution de permanganate de potassium de concentration
0,01mol/L ou 1cm d’une solution de permanganate de potassium de concentration 1mol/L, le flux
émergent ne sera pas le même. Or pour un domaine de concentration donné, Auguste Beer a
montré qu’il existe une relation de proportionnalité entre l’absorbance et la concentration. Aux
vues de ces considérations, on peut modifier le résultat du paragraphe précédent pour y faire
intervenir à la fois le coefficient d’extinction molaire et la concentration. On obtient alors en posant
k′ = ϵc :
 | (4) |
Cette relation constitue la loi de Beer-Lambert. Cette loi reste valable pour :
- Des concentrations faibles
- Une lumière monochromatique
- Un échantillon homogène non fluorescent
- Un échantillon qui n’est pas le siège d’une réaction photochimique
2.4 Allure des spectres
La loi de Beer-Lambert nous permet de relier l’absorbance à la concentration pour une longueur d’onde
donnée. Les spectromètres, eux, fournissent un graphe indiquant l’absorbance ou la transmittance en fonction
de la longueur d’onde. On se propose ici de décrire et de justifier succinctement l’allure de ces
spectres.
Lorsqu’on travaille en phase condensée (liquide) le spectre se présente généralement sous la forme d’une
bande d’absorption en forme de cloche larges et peu nombreuses. Cette allure lisse des spectres en phase
condensée est due à l’interaction intermoléculaire.
Lorsqu’on travaille en phase gazeuse sous faible pression, le spectre est beaucoup plus accidenté, les
interactions moléculaires qui deviennent alors négligeables ne sont plus responsables des bandes d’absorption
et on obtient une ≪ structure fine ≫.
3 Les constituants d’un spectrophotomètre
3.1 Sources
Dans un spectrophotomètre UV/visible, il doit y avoir une source de lumière visible et une source
d’ultraviolet.
La source de lumière visible est généralement une lampe à incandescence avec filament en tungstène et
enveloppe en silice.
La source d’ultraviolet est une lampe à arc au deutérium. Cette source est constituée de deux électrodes
baignant dans une atmosphère de deutérium sous pression réduite. La circulation d’électron entre les deux
électrodes provoque un arc intense. Le choc des électrons sur les molécules de deutérium provoque la
dissociation de celles-ci avec émission de photons. Les photons sont émis suivant un continuum d’émission
dont les longueurs d’onde s’étendent de 160 à 500nm.
3.2 Monochromateur
Dans les spectromètres UV/visibles, l’élément dispersif est un réseau utilisé en réflexion. Dans certains types de spectromètres, ce réseau est au cœur d’un système appelé monochromateur dont la figure 4 illustrele principe.

On se propose ici d’étudier ce système. Pour cela, on assimilera les miroirs C et E à des lentilles L1 et L2 de distances focales respectives f1 et f2. L’axe optique de la lentille L2 forme un angle α avec la normale au réseau. On supposera aussi que les fentes d’entrée et de sortie sont infiniment fines. Enfin, on travaillera en incidence normale. Le schéma correspondant à cette modélisation est représenté sur la figure 5 .

En notant i′ l’angle d’émergence du faisceau, la formule fondamentale des réseaux en incidence normale donne :

Où a est le pas du réseau et k l’ordre d’interférence. Intéressons nous à l’ordre 1. La fente étant supposée infiniment fine, il n’existe qu’une seule longueur d’onde qui peut émerger de ce dispositif par la fente F0. En effet pour obtenir une image au foyer de la lentille L2, les rayons doivent être parallèles à son axe optique. On en déduit alors :

Soit λ0 la longueur d’onde pouvant émerger. L’angle α0 lui correspondant est définit par :

En faisant varier l’angle α, on la radiation émergente ne sera plus λ0 mais une autre radiation de longueur d’onde λn vérifiant la relation :

Le dispositif permet donc de sélectionner dans un spectre large une longueur d’onde bien précise, d’où son nom de monochromateur. Deux points viennent compléter cette étude :
- En réalité les fentes ne sont pas infiniment fine, on n’obtient donc pas en sortie un faisceau
monochromatique mais un faisceau quasi-monochromatique dont les longueurs d’onde sont
situées dans l’intervalle
![[λ0 - Δ λ;λ0 - Δλ ]](Sections/Spectrometrie/UV/Figs/UV16x.png) .
.
- Plus la fente est fine, plus la résolution est meilleure mais il arrive une épaisseur pour laquelle le phénomène de diffraction intervient ce qui explique qu’on ne peut pas indéfiniment affiner la largeur de la fente.
3.3 Détecteurs
Le dernier organe essentiel à tout spectrophotomètre est le détecteur. Dans le cas des spectrophotomètres UV/visible, les détecter les plus courants sont :
- La photodiode(??)
- Le photomultiplicateur (??)
- La barrette de photodiode dans des spectromètres simultanés
4
4 Différents types de spectrophotomètres
4.1 Spectrophotomètres séquentiels
Les spectromètres séquentiels utilisent un monochromateur. On étudie donc longueur d’onde par longueur d’onde l’absorption de l’échantillon. Les détecteurs sont donc des détecteurs de type photodiode ou photomultiplicateurs. On peut rencontrer diverses configurations de spectrophotomètres séquentiels.
4.1.1 Le spectrophotomètre monofaisceau
Utilisés dans bon nombre de dosages de routines, les spectrophotomètres monofaisceaux sont réputés les moins efficaces. Ils ne disposent que d’un seul faisceau et ne peuvent par conséquent réaliser qu’une seule mesure de flux traversant une seule et même cellule à la fois. La détermination de la transmittance nécessitant la comparaison du flux ayant traversé la cellule et le flux avant la cellule, on doit, lorsqu’on utilise ce type d’appareil faire une mesure avec la cellule contenant l’échantillon et une mesure avec une cellule contenant le solvant (le blanc). Ces spectromètres disposent parfois d’une cellule (photodiode) permettant la compensation des variations d’intensité de la source : il s’agit d’une photodiode (5) qui mesure le flux émis par la source et corrige les valeurs d’absorbances (ou transmittance) affichées si celui-ci varie. Le schéma de principe de ces appareils est représenté figure 6 1 .

4.1.2 Le spectrophotomètre à double faisceau
Les spectrophotomètres doubles faisceaux permettent de mesurer simultanément l’absorbance de deux cuves, en l’occurrence :
- L’échantillon dont on veut déterminer l’absorbance
- Le solvant (blanc)
Ces dispositifs possèdent donc un miroir semi réfléchissant permettant de séparer le faisceau lumineux en deux faisceaux d’énergie égale. On peut trouver deux configurations de ces spectrophotomètres :
- Les spectrophotomètres utilisant deux photodiodes, le signal correspond alors proportionnel à la
différences des signaux fournis par les photodiodes(cf. figure 7)
- Les spectrophotomètres utilisant un photomultiplicateur pour détecteur. Dans ces spectrophotomètres,
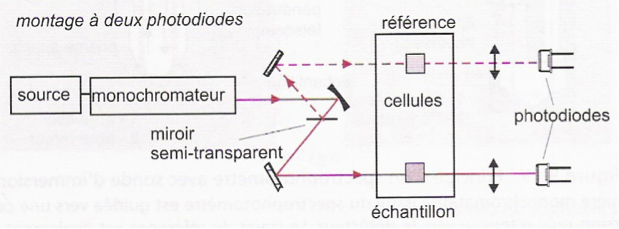
deux miroirs tournant permettent de comparer exactement pour la même longueur d’onde les flux
transmis par l’une ou l’autre des deux voies. Le circuit ajuste la sensibilité du photomultiplicateur en
fonction du flux lumineux qu’il reçoit et en déduit l’absorbance de l’échantillon (cf. figure 8).
4.2 Spectrophotomètres simultanés
Certains spectromètres sont dépourvus de monochromateurs et utilisent en guise de détecteur un détecteur d’images (une barrette de photodiodes). Ces spectrophotomètres très rapides permettent l’analyse de toute une gamme spectrale en un temps record de 1/10ème de secondes. De plus l’absence de monochromateur rend le signal nettement plus lumineux. Le défaut de ces spectromètres réside en la barrette de photodiodes, celles-ci sont miniaturisées au minimum de sorte à ce que la barrette peut en contenir environ 2000, néanmoins leur taille constitue le facteur limitant de la résolution.